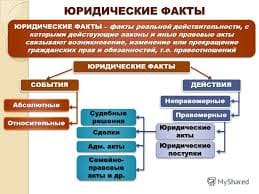
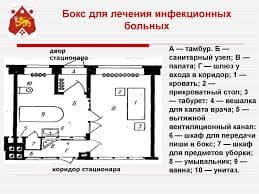
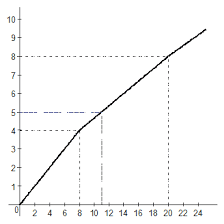
2014-02-09
2014-02-09 1208
12081. Стадия формирования гипертрофии.
Увеличение нагрузки на миокард приводит к усилению интенсивности функционирования структур миокарда, то есть увеличение количества функции, приходящейся на единицу массы сердца.
Если на сердце неожиданно падает большая нагрузка (что при пороках встречается редко), например, при инфаркте миокарда, отрыве сосочковых мышц, разрыве сухожильных хорд, при резком подъеме артериального давления вследствие быстрого увеличения периферического сосудистого сопротивления, то в этих случаях возникает хорошо выраженная кратковременная т.н. "аварийная" фаза первой стадии.
При такой перегрузке сердца количество поступающей в коронарные артерии крови снижается, энергии окислительного фосорлирования для совершения сердечных сокращений не хватает и присоединяется расточительный анаэробный гликолиз. В результате в сердце снижается содержание гликогена, креатин-фосфата, накапливаются недоокисленные продукты (пировиноградная, молочная кислоты), возникает ацидоз, развиваются явления белковой и жировой дистрофии. Повышается содержание натрия в клетках и снижается содержание калия, возникает электрическая нестабильность миокарда, что может провоцировать возникновение аритмии.
Дефицит АТФ ионов калия, ацидоз приводят к тому, что многие медленные кальциевые каналы становятся неактивируемыми при деполяризации и снижается сродство кальция к тропонину, в результате чего клетка сокращается слабее или вообще не сокращается, что может привести к появлению признаков сердечной недостаточности, возникает миогенная дилятация сердца, сопровождающаяся увеличением остающегося во во время систолы в полостях сердца крови и переполнением вен. Повышение давления в полости правого предсердия и в полых венах прямо и рефлекторно вызывает тахикардию, которая усугубляет обменные нарушения в миокарде. Поэтому расширение полостей сердца и тахикардия служат грозными симптомами начинающейся декомпенсации. Если организм не погибает, то очень быстро включается механизм, запускающий гипертрофию: в связи с гиперфункцией сердца, активацией симпатико-адреналовой системы и действием норадреналина на бета-1-адренорецепторы повышается концентрация цАМФ в кардиомиоцитах. Этому же способствует и выход ионов кальция из саркоплазматического ретикулюма. В условиях ацидоза (скрытого или явного) и дефицита энергии усиливается влияние цАМФ на фосфорилирование ядерных энзимных систем, способных увеличить синтез белка, что можно зарегистрировать уже через час после перегрузки сердца. Причем в начале гипертрофии имеет место опережающее усиление синтеза белков митохондрий. Благодаря этому клетки обеспечивают себя энергией для продолжения функции в трудных условиях перегрузки и для синтеза других белков, в том числе и сократительных.
Прирост массы миокарда идет интенсивно, скорость его равна 1 мг/г массы сердца в час. (Например, после отрыва створки аортального клапана у человека масса сердца увеличилась в 2,5 раза за две недели). Процесс гипертрофии продолжается до тех пор, пока интенсивность функционирования структур не нормализуется, то есть пока масса миокарда не придет в соответствие с увеличенной нагрузкой и исчезнет стимул, ее вызвавший.
При постепенном формировании порока эта стадия значительно растягивается во времени. Она развивается медленно, без "аварийной" фазы, исподволь, но с включением тех же механизмов.
Следует подчеркнуть, что формирование гипертрофии находится в прямой зависимости от нервных и гуморальных влияний. Она развивается при обязательном участии соматотропина и вагусных влияний. Существенное положительное влияние на процесс гипертрофии оказывают катехоламины, которые через цАМФ индуцируют синтез нуклеиновых кислот и белков. Инсулин, тиреоидные гормоны, андрогены также способствуют синтезу белков. Глюкокортикоиды усиливают распад белков в организме (но не в сердце или мозге), создают фонд свободных аминокислот и тем самым обеспечивают ресинтез белков в миокарде.
Активируя К-Nа-АТФ-азу, они способствуют поддержанию оптимального уровня ионов калия и натрия, воды в клетках, сохраняют их возбудимость.
Итак гипертрофия закончилась и наступает II стадия ее течения.
II-ая стадия - стадия завершившейся гипертрофии.
В эту стадию наблюдается относительно устойчивая адаптация сердца к непрерывной нагрузке. Процесс потребления АТФ на единицу массы снижается, восстанавливаются энергетические ресурсы миокарда, исчезают явления дистрофии. Интенсивность функционирования структур нормализуется, в то время как работа сердца и, следовательно, потребление кислорода остаются повышенными. Само увеличение толщины стенки создает затруднения для растяжения камеры сердца в период диастолы. Из-за гипертрофии снижается плотность входящего кальциевого тока и поэтому ПД, имея нормальную амплитуду, будет восприниматься СПР как сигнал с меньшей амплитудой и, следовательно, в меньшей степни будут активироваться сократительные белки.
В эту стадию нормальная амплитуда силы сокращения сохраняется за счет увеличения длительности сократительного цикла, вследствие удлинения фазы плато потенциала действия, изменения изоферментного состава миозиновой АТФ-азы (с возрастанием доли изофермента V3, обеспечивающего самый медленный гидролиз АТФ), в результате снижается скорость укорочения миокардиальных волокон и увеличивается длительность сократительного ответа, способствуя поддержанию силы сокращения на обычном уровне, несмотря на уменьшение развития силы сокращения.
Менее благоприятно развивается гипертрофия в детском возрасте, так как рост специализированной проводящей системы сердца отстает от роста его массы по мере прогрессирования гипертрофии.
При устранении препятствия, вызвавшего гипертрофию (операция), происходит полная регрессия гипертрофических изменений в миокарде желудочков, однако сократимость обычно полностью не восстанавливается. Последнее может быть связано с тем, что изменения, происходящие в содинительной ткани (накопление коллагена) не подвергаются обратному развитию. Будет ли регрессия полной или частичной, зависит от степени гипертрофии, а также от возраста и состояния здоровья больного. Если сердце гипертрофировано умеренно, оно может долгие годы работать в режиме компенсаторной гиперфункции и обеспечивать активную жизнь человека. Если же гипертрофия прогрессирует и масса сердца достигает 550 г и более (может достигнуть и 1000 г при норме 200 - 300 г), то в
этом случае все более проявляется действие неблагоприятных факторов, которые в конце концов приводят к "отрицанию отрицания", то есть к изнашиванию миокарда и наступлению III-ей стадии течения гипертрофии.
Факторы, влияющие на сердце неблагоприятно и вызывающие "изнашивание" миокарда:
1. При патологической гипертрофии ее формирование происходит на фоне сниженного или неизмененного минутного объема, то есть количество крови, приходящееся на единицу массы миокарда, снижается.
2. Увеличение массы мышечных волокон не сопровождается адекватным увеличением количества капилляров (хотя они и шире обычных), плотность капиллярной сети значительно снижается. Например, в норме приходится 4 тыс. капилляров на 1 мкм, при патологической гипертрофии 2400.
3. В связи с гипертрофией снижается плотность иннервации, снижается концентрация норадреналина в миокарде (в 3 - 6 раз), снижается реактивность клеток к катехоламинам в связи с уменьшением площади адренорецепторов. Это приводит к уменьшению силы и скорости сердечных сокращений, скорости и полноты диастолы, снижению стимула к синтезу нуклеиновых кислот, поэтому ускоряется изнашивание миокарда.
4. Увеличение массы сердца происходит за счет утолщения каждого кардиомиоцита. Объем клетки при этом увеличивается в большей степени, чем площадь поверхности, несмотря на компенсаторные изменения в сарколемме (увеличение количества Т-тубул), то есть уменьшается отношение поверхности к объему. В норме оно равно 1:2, а при выраженной гипертрофии 1:5. В результате поступления глюкозы, кислорода и других энергетических субстратов на единицу массы снижается, снижается и плотность входящего кальциевого тока, что способствует снижению силы сердечных сокращений.
5. В силу тех же причин снижается отношение рабочей поверхности СПР к массе саркоплазмы, что приводит к снижению эффективности кальциевого "насоса", СПР и часть ионов кальция не откачивается в продольные цистерны СПР).
Избыток кальция в саркоплазме приводит:
1) к контрактуре миофибрилл
2) падению эффективности использования кислорода из-за действия
избытка кальция на митохондрии (см. раздел "Повреждение клетки")
3) активируются фосфолипазы и протеазы, которые усугубляют повреждение клеток вплоть до их гибели.
Таким образом, по мере прогрессирования гипертрофии все больше нарушается использование энергии. При этом, наряду с плохой сократимостью наблюдается затруднение расслабления мышечного волокна, возникновение локальных контрактур, а в дальнейшем - дистрофия и гибель кардиомиоцитов. Это увеличивает нагрузку на оставшиеся, что приводит к изнашиванию генераторов энергии - митохондрий и еще более выраженному снижению силы сердечных сокращений.
Таким образом, прогрессирует кардиосклероз. Оставшиеся клетки не могут справиться с нагрузкой, развивается сердечная недостаточность. Следует отметить, что и наличие компенсаторной физиологической гипертрофии снижает устойчивость организма к различным видам гипоксии, длительным физическим и психическим нагрузкам.
При снижении функциональных способностей миокарда включаются и экстракардиальные механизмы компенсации. Основная их задача - привести кровообращение в соответствие с возможностями миокарда.
Первая группа таких механизмов - это кардиоваскулярные (сердечно-сосудистые) и ангиоваскулярные (сосуд-сосудистые) рефлексы.
1. Депрессорно-разгрузочный рефлекс. Он возникает в ответ на повышение давления в полости левого желудочка, например, при стенозе устья аорты. При этом усиливается афферентная импульсация по блуждающим нервам и рефлекторно снижается тонус симпатических нервов, что приводит к расширению артериол и вен большого круга. В результате уменьшения периферического сосудистого сопротивления (ПСС) и снижения венозного возврата к сердцу происходит разгрузка сердца.
Одновременно возникает брадикардия, удлиняется период диастолы и улучшается кровоснабжение миокарда.
2. Рефлекс, противоположный предыдущему - прессорный, возникает в ответ на понижение давления в аорте и левом желудочке. В ответ на возбуждение барорецепторов сино-каротидной зоны, дуги аорты возникает сужение артериальных и венозных сосудов, тахикардия, то есть в этом случае снижение минутного объема компенсируется уменьшением емкости периферического сосудистого русла,
что позволяет поддерживать артериальное давление (АД) на адекватном уровне. Так как эта реакция не касается сосудов сердца, а сосуды головного мозга даже расширяются, то их кровоснабжение страдает в меньшей степени.
3. Рефлекс Китаева. (См. лекцию ВСО N2)
4. Разгрузочный рефлекс В.В.Парина - трехкомпонентный: брадикардия, снижение ПСС и венозного возврата.
Включение этих рефлексов приводит к уменьшению минутного объема, но уменьшает опасности отека легких (то есть развитию острой сердечной недостаточности (ОСД)).
Вторая группа экстракардиальных механизмов - компенсаторные изменения диуреза:
1. Активация ренин-ангиотензиновой системы (РАС) в ответ на гиповолемию приводит к задержке соли и воды почками, что приводит к увеличению объема циркулирующей крови, что вносит определенный вклад в поддержание минутного объема сердца.
2. Активация натрийуреза в ответ на повышение давления в предсердиях и секрецию натрийуретического гормона, что способствует снижению ПСС.
* * *
Если компенсация с помощью выше разобранных механизмов оказывается несовершенной, возникает циркуляторная гипоксия и вступает в действие третья группа экстракардиальных компенсаторных механизмов, о которых говорилось в лекции по дыханию, в разделе "Приспособительные механизмы при гипоксиях".