 2015-04-23
2015-04-23 1493
1493Алкени в природі зустрічаються рідко. Нижчі алкени можуть входити до складу нафтового газу, вищі – деяких нафт. Найважливіший промисловий постачальник ненасичених вуглеводнів є нафтопереробна промисловість. Великі кількості алкенів і алкінів утворюються при крекінгу і піролізі нафти.
Піроліз – найефективніший сучасний промисловий спосіб добування нижчих алкенів. Структура продуктів і їхні виходи залежать від сировини й умов процесу. Гази, що виходять у кількості до 25% від сировини, багаті газоподібними алкенами С2-С4. Крекінг-бензини містять багато рідких алкенів.
Іншим важливим промисловим способом одержання алкенів є дегідрування алканів (каталізатор – хром (III) оксид, температура 450-4600С):
(СН3)2СН – СН3 ® (СН3)2С = СН2 + Н2.
Лабораторні способи одержання алкенів здебільшого є реакціями відщеплення. Найважливіший з цих способів – дегідратація спиртів. При нагріванні спиртів з водовід¢ємними речовинами (концентрована сульфатна чи фосфатна кислота) при підвищеній температурі йде відщіплення води. З етилового спирту одержують етилен:
СН3–СН2ОН ® СН2=СН2 + Н2О.
При дегідратації спиртів Гідроген переважно відщеплюється за правилом А. М. Зайцева (1875 р.) – від того із сусідніх атомів Карбону, що бідніший Гідрогеном:
 СН3–СН2–СН–СН3 ® СН3–СН=СН–СН3 + Н2О.
СН3–СН2–СН–СН3 ® СН3–СН=СН–СН3 + Н2О.
ОН
При нагріванні алкілгалогенідів з концентрованими спиртовими розчинами лугів відбувається відщеплення галогеноводню – дегідрогалогенування (також за правилом Зайцева). Легше всього реакція йде з третинними галогенопохідними, важче – з первинними.


 СН3 – СН2 – СН – СНІ – СН3 КОН(спирт. р-н) – НI СН3 – СН2 – С = СН – СН3
СН3 – СН2 – СН – СНІ – СН3 КОН(спирт. р-н) – НI СН3 – СН2 – С = СН – СН3
СН3 СН3
чи


 СН3 – СН – СН – СН – СН3 цинковий пил СН3 – СН = СН – СН – СН3 + Br2.
СН3 – СН – СН – СН – СН3 цинковий пил СН3 – СН = СН – СН – СН3 + Br2.
Br Br СН3 СН3
(використовуються віцинальні дигалогенопохідні, коли атоми галогенів знаходяться біля сусідніх атомів Карбону).
3. Алкени за фізичними властивостями близькі до алканів. Однак температури кипіння їх трохи нижче, ніж у відповідних алканів, а густина трохи вище. Перші три члени ряду С2-С4 – гази, С5-С17 – рідини, вище – тверді речовини. Ізомерія карбонового ланцюга, положення подвійного зв'язку, а також просторова конфігурація відбиваються на фізичних константах ізомерів.
Всі алкени мають густину менше за одиницю, мають характерний запах, у воді розчинні погано, але все-таки краще, ніж відповідні алкани.
Хімічна поведінка алкенів визначається наявністю подвійного зв'язку. Електронна густина s-зв'язку концентрується по лінії, що з'єднує ядра атомів, а електронна густина p-зв'язку виходить за ці межі і утворює більш велику область негативного заряду. Характерна риса p-електронів – їхня рухливість, вони менш міцно зв¢язані з ядрами атомів, чим s-електрони. Тому p-зв'язок легше поляризується. Електрони p-зв'язку легко вступають у хімічні реакції з електрофільними реагентами, що викликають їх гетеролітичний розрив. Енергія утворення подвійного зв'язку складає 620 кДж/моль, а простого – 348 кДж/моль. Їхня різниця (близько 270 кДж/моль) характеризує міру міцності p-зв'язку. При руйнуванні p-зв'язку відбуваються реакції приєднання з утворенням двох нових s-зв'язків. Такі реакції типові для алкенів.
Реакції електрофільного приєднання до подвійного зв'язку відбуваються по гетеролітичному типу і є ступінчастим іонним процесом. Вирішальною стадією таких реакцій є взаємодія електрофільного реагенту з p-електронною хмарою подвійного зв'язку. Часто в ролі електрофільного реагенту виступає протон Н+, що утворюється при дисоціації мінеральних кислот (НС1, НВг, НІ, Н2SО4). Спочатку електрофільный реагент за рахунок свого позитивного заряду взаємодіє з p-електронами подвійного зв'язку, утворюючи p-комплекс. Потім з одним з ненасичених С-атомів за участю р-електронів подвійного зв'язку утворюється звичайний s-зв'язок. Сусідній атом Карбону – другий партнер подвійного зв'язку, позбавивши електрона, здобуває позитивний заряд:



 Н+ Н
Н+ Н

 С = С + Н+ С = С – С – С+ –
С = С + Н+ С = С – С – С+ –
p-комплекс карбокатіон
В другій, завершальній стадії карбокатіон реагує з аніоном Х–, утворюючи другий s-зв'язок. У результаті виходить кінцевий продукт приєднання:

 Н Н Х
Н Н Х
 – С – С+ – + Х– – С – С –
– С – С+ – + Х– – С – С –
Звичайно перша стадія йде повільніше, ніж друга.
1. Галогенгідрування (приєднання галогеноводнів). Як приклад розберемо приєднання бромоводню до бутену-2:
 НВr ® Н+ + Вr– Н+
НВr ® Н+ + Вr– Н+
СН3 – СН = СН – СН3 + Н+ ® СН3 – СН = СН – СН3 ®
 ® СН3 – СН2 – СН+ – СН3 + Вr– СН3 – СН2 – СНВr – СН3.
® СН3 – СН2 – СН+ – СН3 + Вr– СН3 – СН2 – СНВr – СН3.
Приєднання гідрогенброміду протікає як двухстадійний іонний процес. Вирішальна стадія всієї реакції – приєднання протона з утворенням карбокатіона. Це доводиться тим фактом, що приєднуватися здатні галогеноводні, але не їхнї солі.
У реакціях приєднання несиметричних реагентів типу НХ до несиметрично побудованих алкенів, наприклад R– СН=СН2, виникає питання про порядок приєднання. Закономірності подібних реакцій були вивчені В. В. Марковніковим (1838-1904 р.) і привели до формулювання правила Марковнікова (1869 р.): при приєднанні галогеноводнів до несиметричних алкенів Гідроген приєднується до більш гідрогенізованого атома Карбону, а галоген – до менш гідрогенізованого:
СН3 – СН = СН2 + НВr ® СН3 – СНВr – СН3.
Правило Марковнікова знаходить пояснення з позицій електронної теорії органічних реакцій. Атом Карбону в СН3-групі несе частковий негативний заряд і внаслідок ефекту гіперконьюгації уздовж ланцюга спостерігається чергування позитивних і негативних зарядів:

 Н
Н
 Н ®С ® СН = СН2
Н ®С ® СН = СН2
Н
Протон приєднується до того ненасиченого атома Карбону, що несе частковий негативний заряд (це атом, більш насичений Гідрогеном). Аніонна частина реагенту направляється до найменш гідрогенізованого. Правило Марковнікова діє тільки при іонному механізмі приєднання. У загальному виді його можна сформулювати так: електропозитивна (катіонна) частка реагенту приєднується до атома Карбону, що містить більше число атомів Гідрогену, а електронегативна (аніонна) – до атома Карбону з меншим числом атомів Гідрогену. Хоча порядок приєднання обумовлений у першу чергу будовою молекули алкена, він залежить також від умов реакції. Зокрема, у присутності пероксидів реакція здобуває радикальний характер і приєднання йде проти правила Марковнікова.
2. Галогенування. За електрофільним механізмом йде і приєднання галогенів. Під впливом електронної густини подвійного зв'язку відбувається поляризація в молекулі галогену, тобто електрофільність симетричної молекули галогену може проявитися тільки після поляризації. Далі реакція йде за описаним вище шляхом:
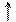 Вr2 ® Вrd+ – Вrd– Вrd+
Вr2 ® Вrd+ – Вrd– Вrd+
СН3 – СН = СН – СН3 + Br+ ® СН3 – СН = СН – СН3 ®
 ® СН3 – СНВr – СН+ – СН3 + Вr– СН3 – СНВr – СНВr – СН3.
® СН3 – СНВr – СН+ – СН3 + Вr– СН3 – СНВr – СНВr – СН3.
Алкени за звичайних умов приєднують галогени, особливо легко хлор і бром. У результаті утворюються дигалогенопохідні алканів. Знебарвлення бромної води є якісною реакцією на подвійний зв'язок. Для цього застосовують розчин брому в чьотирьоххлористому карбоні чи хлороформі.
3. Гідратацію – приєднання води до алкенів (пряму гідратацію) проводять у присутності фосфатних каталізаторів (Н3РО4 на алюмосилікаті із солями кадмію, купруму, кобальту; 3000С; 8 МПа):
 СН2 = СН2 + Н2О kat, Р, Т СН3 – СН2ОН.
СН2 = СН2 + Н2О kat, Р, Т СН3 – СН2ОН.
4. Гідрування – приєднання водню здійснюється в присутності каталізаторів, що атомізують молекули водню, тобто здійснюється каталітичне гідрування. Найбільш активними каталізаторами є Платина, Паладій, на практиці частіше використовують мілкороздрібнений нікель. Особливо активну форму нікелевого каталізатора являє собою «нікель Ренея», що називають також скелетним нікелем:
 СН2 = СН2 + Н2 kat, СН3 – СН2ОН.
СН2 = СН2 + Н2 kat, СН3 – СН2ОН.
5. Реакції полімеризації. Полімеризація – одна з найбільш важливих у промисловому відношенні реакція алкенів. Продукти полімеризації нижчих алкенів – етилену, пропілену, ізобутену – використовують для виробництва пластичних мас, синтетичних волокон і інших практично важливих матеріалів. Здійснити полімеризацію алкенів з утворенням твердих полімерних продуктів змогли лише в 40-50-х роках нашого сторіччя. Найпростіший з алкенів – етилен можна полімеризувати різними способами: під тиском 120-150 МПа, ініціюючи полімеризацію киснем чи органічними пероксидами (поліетилен високого тиску); при низькому тиску на металорганічних каталізаторах – сумішах алюмінійалкилів із солями титану (поліетилен низького тиску).
СН2 = СН2 + СН2=СН2 + … ® –СН2 – СН2 – СН2 – СН2– ® (-СН2 – СН2 –)n
поліетилен
Поліетилен – насичений вуглеводень з молекулярною масою від 20 тис. до 1 млн. Являє собою прозорий матеріал, що має високу хімічну стійкість; температура розм'якшення 100-1300С; напруження руйнування при розтяганні складає 12-34 МПа, має низьку тепло- і електропровідності. Поліетилен застосовують для ізоляції електричних проводів, виготовлення прозорих плівок (пакувальний матеріал), замість стекол для укриття рослин у парниках. З поліетилену роблять посуд і інші вироби.
Поліпропілен одержують із пропілену аналогічно поліетилену. Довгий час вважали, що при полімеризації пропілену можна отримати лише олієподібні продукти. Коли ж навчилися проводити стереоспецифічну полімеризацію пропілену, виявилося, що при цьому виходить прозорий матеріал з температурою розм'якшення 160-1700С (руйнівне напруження при розтяганні 26-35 МПа), що має гарні електроізоляційні властивості. Продавлюючи розплав поліпропілену через фільєри, одержують нитки поліпропіленового волокна, що має велику міцність і хімічну стійкість. З нього виготовляють канати, рибальські мережі, фільтрувальні тканини.
Сутність стереоспецифічної полімеризації пропілену полягає в створенні строго упорядкованої просторової структури лінійного ланцюга полімеру. При полімеризації в ланцюзі полімеру виникають асиметричні атоми Карбону. Їхня конфігурація може чергуватися безладно; це характерно для атактичного поліпропілену. При стереоспецифічній полімеризації виникають просторово упорядковані структури. У ізотактичному полімері всі однакові групи –СН3 знаходяться по одну сторону площини молекули, а всі атоми Н – по іншу. Саме ізотактичні стереорегулярні полімери мають найбільш цінні фізико-механічні властивості.
6. Окиснення. Алкени легко піддаються дії різних окисників, різко відрізняючись в цьому відношенні від алканів і циклоалканів. У залежності від умов окиснення утворюються різні продукти. У самих жорстких умовах, при спалюванні на повітрі, алкени перетворюються в СО2 і воду:
С2Н4 + 3О2 —> 2СО2 + 2Н2О.
У більш м'яких умовах окиснення в першу чергу йде по подвійному зв'язку. Обережне окиснення лужним розчином перманганату калію приводить до утворення двохатомних спиртів – гліколей:
 С2Н4 КМпО4 НО-СН2-СН2-ОН
С2Н4 КМпО4 НО-СН2-СН2-ОН
Реакція (російський хімік Е. Е. Вагнер) протікає швидко, на холоду, причому спостерігається знебарвлення характерного кольору перманганату. Це якісна проба на подвійний зв'язок.
При дії більш енергійних окиснювачів (кислотний розчин перманганату, хромова суміш) відбувається окисне розщеплення молекули алкена по подвійному зв'язку. Як приклад наведемо реакції окиснювання трьох ізомерних бутенів:
СН3 – СН2 – СН = СН2 + [O] ® СН3 – СН2 – СООН + CO2;
бутен-1 пропіонова кислота
СН3 – СН = СН – СН3 + [O] ® 2СН3 – СООН;
бутен-2 ацетатна кислота
 СН3 – С = СН2 + [O] ® (СН3)2– С = О + СО2 + Н2О.
СН3 – С = СН2 + [O] ® (СН3)2– С = О + СО2 + Н2О.
СН3 ацетон
2-метилпропен (ізобутен)
Продукти окиснення дозволяють зробити висновок про положення подвійного зв'язку в молекулі, про будову карбонового скелета.
Каталітичне окиснення киснем повітря приводить до утворення a-оксидів, що мають велике значення в органічному синтезі. Так, наприклад, одержують дуже важливий технічний продукт — етиленоксид:
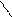
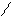 О
О
 СН2 = СН2 + О2 Аg, нагрівання Н2С –– СН2.
СН2 = СН2 + О2 Аg, нагрівання Н2С –– СН2.
оксид етилену, етиленоксид
Алкени легко реагують з озоном, утворюючи продукти приєднання – циклічні пероксиди – озоніди (в¢язкі рідини чи тверді речовини, дуже нестійкі, легко вибухають):
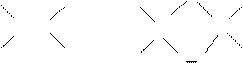 О
О
С = С + О3 ® С С
О О
7. Реакції заміщення. У деяких випадках алкени можуть вступати у реакції заміщення. Найбільш легко заміщаються Гідроген у a-карбонового атома Карбону відносно подвійного зв'язку:
СН3– СН = СН2 + С12 ® СН2С1– СН = СН2 + НС1.
3-хлор-пропен-1
Застосування. Алкени широко використовують в хімічній промисловості як сировину для отримання різноманітних органічних речовин і матеріалів. Етен – початкова речовина для виробництва етанолу, етилгліколю, епоксидів, дихлоретану. Велика кількість етену переробляється в поліетилен, який використовується для виготовлення пакувальної плівки, посуду, труб, електроізоляційних матеріалів, медичної апаратури.
З пропену отримують гліцерол, ацетон, ізопропанол, розчинники. Полімеризацією пропену отримують поліпропілен, який по багатьом показникам перевершує поліетилен: має вищу температуру плавлення, хімічну стійкість. З полімерів – аналогів поліетилену одержують волокна, що володіють унікальними властивостями. Так, наприклад, волокно з поліпропілену прочніше всіх відомих синтетичних волокон. Матеріали, виготовлені з цих волокон, є перспективними і знаходять все більше застосування в різних областях людської діяльності, особливо в медицині.
Лекція № 4. Ароматичні сполуки (арени)
План
1. Будова ароматичних вуглеводнів.
2. Фізичні та хімічні властивості аренів.
1. Найпростішим представником одноядерних ароматичних вуглеводнів є бензен С6Н6. Вперше бензен був здобутий М. Фарадеем у 1825 р. з конденсованих залишків світильного газу, який утворюється в процесах переробки кам’яного вугілля. 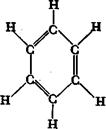 У 1865 р. А. Кекулє запропонував формулу у вигляді циклу з шести атомів Карбону з розташованими по черзі простими та подвійними зв’язками. Пізніше було доведено, що ці зв’язки рівноцінні (А. Ладенбург, 1874 р. та Е. А. Врублевський, 1878 р.). У відповідності до сучасних уявлень, основаних на даних квантової хімії та фізико-хімічних досліджень, молекула бензену являє собою правильний плоский шестикутник. Усі карбонові атоми знаходяться у стані sр 2-гібридізації. При цьому кожний атом Карбону утворює три s-зв’язки (один С – Н два С – С), які лежать в одній площині під кутом 120° один до одного, і надає одну р -орбіталь для утворення замкненої спряженої системи.
У 1865 р. А. Кекулє запропонував формулу у вигляді циклу з шести атомів Карбону з розташованими по черзі простими та подвійними зв’язками. Пізніше було доведено, що ці зв’язки рівноцінні (А. Ладенбург, 1874 р. та Е. А. Врублевський, 1878 р.). У відповідності до сучасних уявлень, основаних на даних квантової хімії та фізико-хімічних досліджень, молекула бензену являє собою правильний плоский шестикутник. Усі карбонові атоми знаходяться у стані sр 2-гібридізації. При цьому кожний атом Карбону утворює три s-зв’язки (один С – Н два С – С), які лежать в одній площині під кутом 120° один до одного, і надає одну р -орбіталь для утворення замкненої спряженої системи.
Сукупність специфічних властивостей бензену, а саме висока стабільність, інертність в реакціях приєднання та схильність до реакцій заміщення, одержала назву ароматичність, або ароматичні властивості.
У 1931 р. німецький вчений Е. Хюккель на підставі квантово-хімічних розрахунків за допомогою методу МО сформулював правило стабільності циклічних спряжених систем, котре являє собою теоретично обґрунтований метод визначення їх ароматичності. Критерієм ароматичності органічної сполуки є наявність в його структурі плоского циклу, що вміщує замкнену спряжену систему, яка включає (4 n + 2) p-електронів, де n = 0, 1, 2, 3 і т. д. До найбільш поширених ароматичних систем, які містять 6 p-електронів (n = 1), відноситься бензен і його похідні. Правило Хюккеля застосовується і до систем з конденсованими ядрами, такими, як нафталін, антрацен і фенантрен:
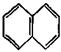
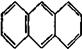
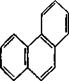
нафталін 10p-електронів антрацен 14p-електронів фенантрен 14p- електронів
(n =2) (n =3) (n =3)
2. Фізичні властивості.За звичайних умов бензен і його ближчі гомологи є рідинами і мають специфічний запах. Усі арени не розчинні у воді і добре розчиняються в органічних розчинниках, є розчинниками для багатьох органічних речовин. Через високий вміст атомів Карбону згоряють з сильно коптячим полум’ям. Арени є дуже отруйними. Вдихання їх парів викликає лейкемію. Властивості деяких аренів надані в таблиці.
| Назва | Температура, °С | d 420 | |
| плавлення | кипіння | ||
| Бензен | 5,5 | 80,1 | 0,8790 |
| Толуєн | –95,0 | 110,6 | 0,8669 |
| о -Ксилол | –25,0 | 144,4 | 0,8802 |
| м -Ксилол | 47,9 | 139,1 | 0,8641 |
| Стирол | –30,6 | 145,2 | 0,9060 |
Хімічні властивості. Незважаючи на те, що за формулою бензен мусить бути ненасиченою сполукою, для нього і його гомологів та для інших аренів більш характерні реакції заміщення, а не приєднання (як для олефінів, алкінів, тощо). Реакційна здатність бензену і його гомологів визначається головним чином наявністю в структурі замкненої p-електронної системи, яка є областю підвищеної електронної густини молекули і здатна притягувати позитивно заряджені частинки – електрофіли. Тому ароматичні вуглеводні, як і алкени, мають нуклеофільний характер. Проте арени, на відміну від ненасичених сполук, при взаємодії з електрофільними реагентами більше схильні не до реакцій приєднання, а до реакцій заміщення, оскільки при цьому зберігається їх ароматична система. Ці реакції носять назву реакцій електрофільного заміщення SE. Реакції приєднання для аренів менш характерні, бо вони призводять до порушення ароматичності, дуже важко вступають ароматичні вуглеводні і в реакції окиснення.
І. Реакції електрофільного заміщення (SE)
Бензен і його гомологи порівняно легко вступають у реакції електрофільного заміщення. Електрофільна частинка, що атакує p-електронну систему бензенового кільця, може являти собою позитивно заряджений іон Е + або частину нейтральної молекули, яка має центр зі зменшеною електронною густиною Е d+® Х d-. Утворення електрофільних частинок для участі в реакції можливе різними способами – під дією p-електронної системи бензенового кільця, каталізатора, розчинника та ін. Незважаючи на велике різноманіття електрофільних реагентів і ароматичних систем, переважна більшість реакцій електрофільного заміщення в ароматичному ряді описується в рамках єдиного загального механізму.
При атаці електрофільною частинкою спочатку утворюється нестійкий p-комплекс. Ця стадія реакції є швидкою та оборотною. При цьому ароматичність не втрачається. Далі p-комплекс перетворюється на s-комплекс (карбокатіон), де електрофільна частинка утворює ковалентний зв’язок з одним атомом Карбону – він переходить в стан sр 3-гібридізації і ароматичність втрачається. Ця стадія є найбільш високоенергетичною, вона визначає швидкість процесу в цілому. Перебіг електрофільного заміщення через стадію утворення s-комплексу підтверджено численними дослідженнями. В деяких випадках s-комплекс удалося виявити спектральними методами і навіть виділити у кристалічному стані. Незважаючи на відносну стабільність s-комплексу за рахунок розподілу позитивного заряду поміж п’ятьма атомами Карбону, він значно менш стійкий, ніж ароматичний секстет. Щоб набути більшої стабільності s-комплекс відщеплює протон від атома Карбону, зв’язаного з електрофілом і через те відновлює ароматичність бензенового кільця. Відновлення ароматичної структури дає виграш енергії в 42 кДж/моль.
До найважливіших реакцій електрофільного заміщення в бензольному ядрі відносяться реакції нітрування, галогенування, сульфування, алкілування і ацилування.
1. Нітрування Це процес заміщення атома водню в бензеновому ядрі на нітрогрупу–NО2. Нітруючими реагентами в реакції є концентрована нітратна кислота або суміш концентрованих нітратної та сульфатної кислот (нітруюча суміш). З HNO3 конц бензен і його гомологи реагують повільно. Тому для нітрування аренів найчастіше застосовується нітруюча суміш:
 С6Н6 + HNO3 H2SO4 C6H5NO2 + H2O.
С6Н6 + HNO3 H2SO4 C6H5NO2 + H2O.
Атакуючою електрофільною частинкою в реакції нітрування є іон нітронію N02+ котрий утворюється в результаті кислотно-основної реакції між кислотами, де нітратна кислота відіграє роль основи:
OH–NO2 + H2SO4 ® NO2+ + HSO4– + H2O
іон нітронію
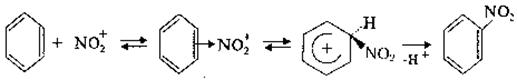 Іон нітронію атакує p-електронну систему бензенового ядра, утворюючи нітропохідне:
Іон нітронію атакує p-електронну систему бензенового ядра, утворюючи нітропохідне:
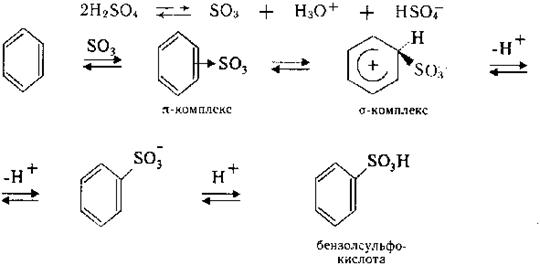
2. Сульфування є процесом заміщення атома Гідрогену в бензольному ядрі на сульфогрупу – SО3Н. В результаті взаємодії утворюються аренсульфокислоти. Сульфування аренів є оборотною реакцією. Вода, яка утворюється в процесі взаємодії, зміщує рівновагу вліво. Тому для збільшення виходу цільового продукту беруть надлишок концентрованої сульфатної кислоти або використовують олеум (розчин триоксиду сірки SО3 в сульфатній кислоті). Атакуючою електрофільною частинкою в реакції служить триоксид сірки SО3:
3. Галогенування. 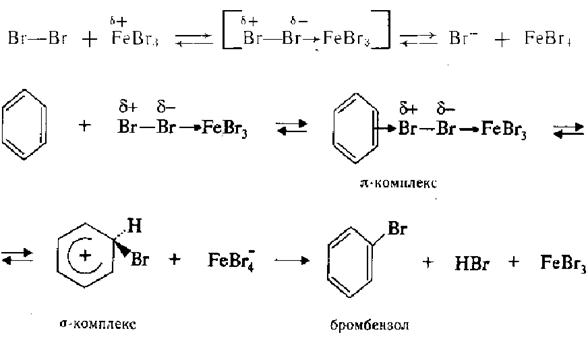 Заміщення атома водню в бензольному ядрі на атом хлору або брому здійснюють дією вільного хлору або брому в присутності каталізаторів — кислот Льюїса (А1С13, FеВг3, та ін.):
Заміщення атома водню в бензольному ядрі на атом хлору або брому здійснюють дією вільного хлору або брому в присутності каталізаторів — кислот Льюїса (А1С13, FеВг3, та ін.):
Під дією каталізатора, на атомі металу котрого є дефіцит електронної густини, молекула галогену поляризується. Атакуючою електрофільною частинкою в цьому випадку служить або комплекс поляризованої молекули галогену з кислотою Льюїса, або катіон галогену, що утворюється в процесі іонізації даного комплексу:
4. Алкілування за Фріделем – Крафтсом
Для введення алкільної групи в молекулу бензену та його гомологів як електрофільні реагенти найчастіше використовують галогеналкани. Взаємодія аренів з галогеналканами відбувається в присутності каталізаторів – кислот Льюїса (А1С13, FеС13, ZnС12, ВF3, SnС12 та ін.), з яких найчастіше застосовують хлорид алюмінію:
 C6H6 + C2H5Cl AlCl3 C6H5C2H5 + HCl
C6H6 + C2H5Cl AlCl3 C6H5C2H5 + HCl
Крім галогеналканів для алкілування аренів можуть використовуватися спирти і алкени. Реакції за участю спиртів проходять в присутності кислот Льюїса або мінеральних кислот (Н3РО4, Н2SO4):
Алкілування аренів алкенами вимагає присутності як каталізатора кислот Льюїса та мінеральної кислоти в ролі джерела протонів.
Атакуючою електрофільною частинкою в реакції алкілування за Фріделем – Крафтсом є карбкатіон, котрий утворюється в кожному конкретному випадку. 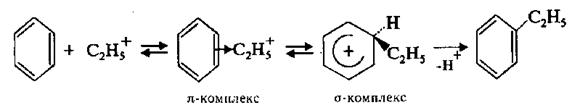 За своїм механізмом реакція алкілування аналогічна розглянутим вище реакціям нітрування, сульфування та галогенування:
За своїм механізмом реакція алкілування аналогічна розглянутим вище реакціям нітрування, сульфування та галогенування:
Введена в бензенове кільце алкільна група, яка є електронодонором, підвищує реакційну здатність ароматичного кільця у відношенні до електрофільних реагентів. Тому утворений в процесі алкілування продукт більше схильний до взаємодії з електрофілом, ніж вихідний арен. У результаті процес алкілування часто не зупиняється на стадії утворення монозаміщеного продукту, а йде далі, даючи ди- та поліалкіларени.
Ацилування – процес введення в молекулу органічної сполуки ацильної групи (R – С=О). Ацилування бензену та його гомологів за Фріделем–Крафтсом звичайно здійснюють галогенангідридами або ангідридами карбонових кислот у присутності кислот Льюїса. Реакція служить загальним методом добування ароматичних кетонів:
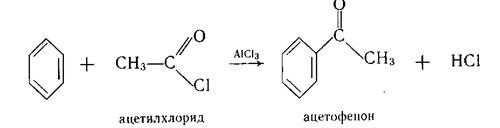
Ацилування ароматичного ядра, на відміну від алкілування, проходить переважно з утворенням монозаміщених продуктів. Ацильна група є електроноакцептором і через це знижує реакційну здатність бензольного кільця у взаємодії з електрофільними реагентами. Тому діацилпохідні утворюються тільки за жорстких умов.
ІІ. Реакції приєднання для ароматичних вуглеводнів не є характерними. Проте за жорстких умов вони все ж відбуваються.
1. Гідрування
 При підвищених температурі та тиску, в присутності каталізаторів, з яких частіше використовують дрібнопористий нікель (нікель Ренея), бензен і його гомологи приєднують три молекули водню, утворюючи циклогексан і його похідні: + 3Н2 ®
При підвищених температурі та тиску, в присутності каталізаторів, з яких частіше використовують дрібнопористий нікель (нікель Ренея), бензен і його гомологи приєднують три молекули водню, утворюючи циклогексан і його похідні: + 3Н2 ®
Останні гідруються значно легше, ніж бензен.
2. Хлорування
При інтенсивному сонячному освітленні або під дією ультрафіолетового випромінювання бензол приєднує хлор. Реакція проходить за радикальним механізмом, з утворенням суміші стереоізомерів гексахлорциклогексану (гексахлорану):
С6Н6 + ЗС12 ® С6Н6СІ6
ІІІ. Реакції окиснення
Окиснення бензенового циклу. Бензенове кільце дуже стійке до дії окисників. За звичайних умов такі сильні окисники, як перманганат калію, нітратна кислота, оксид хрому (VI), пероксид водню та інші, не окиснюють бензен. Але за жорстких умов, наприклад при дії кисню повітря в присутності оксиду ванадію (V) як каталізатора, при температурі 400-5000С бензенове ядро окиснюється, утворюючи малеїновий ангідрид:
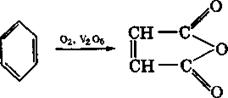
+ 2Н2О + 2СО2
малеїновий ангідрид
Окиснення гомологів Бензену. Алкілбензоли, на відміну від незаміщеного бензолу, окиснюються значно легше. В цьому випадку при дії сильних окисників (КМпО4 та ін.) піддаються окисненню бокові ланцюги. Продуктами окислення є ароматичні карбонові кислоти. Причому кожний алкільний радикал в бензеновому кільці, незалежно від довжини карбонового ланцюга, окиснюється в карбоксильну групу:
ІV. Реакції в боковому ланцюзі
Галогенування гомологів бензену за участю бокового ланцюга.
В умовах вільнорадикального заміщення (при дії світла) відбувається заміщення в боковому ланцюзі:
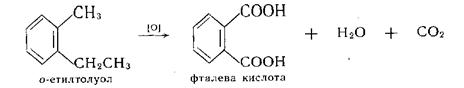
Застосування. Ароматичні вуглеводні широко застосовують у виробництві барвників, пластичних мас, хіміко-фармацевтичних препаратів, вибухових речовин.

