 2015-07-04
2015-07-04 2839
2839Основным способом решения уравнений Хартри-Фока в квантовой химии на сегодня является метод Рутана, в котором молекулярные орбитали ищутся в виде конечных разложений по системе базисных функций, центрированных на атомах (подход МО ЛКАО).
Если в пределе бесконечного базиса (полной системы функций) выбор конкретного вида базисных функций не имеет значения (получается точный результат, называемый хартри-фоковским пределом), то для используемых на практике конечных разложений выбор базиса становится критическим шагом, определяющим качество результатов, с одной стороны и вычислительные затраты с другой стороны.
В связи с этим приобретает актуальность выяснение сходимости результатов по отношению к базису с целью нахождения базиса, обеспечивающего адекватное описание изучаемых систем при приемлемых затратах времени на вычисления. При этом обычно последовательно улучшают базис путем его расширения, останавливаясь там, где результаты перестают меняться ("сходятся") в пределах точности, которая достаточна для корректного решения задачи.
Сравнительный анализ базисов и результатов является важным практическим аспектом, определяющим успех любого проекта, связанного с использованием неэмпирических квантовохимических расчетов.
Слэтеровские и гауссовы базисы.
В настоящее время в качестве базисных функций, по которым производится разложение в методе самосогласованного поля Хартри-Фока-Рутана, используются преимущественно гауссовы орбитали. Для этих же целей могут использоваться также слэтеровские орбитали, которые теперь однако почти полностью утратили свое практическое значение. Дело в том, что не смотря на определенные преимущества, слэтеровские орбитали применимы в молекулярных расчетах лишь для двухатомных и линейных молекул. В остальных случаях вычисление двухэлектронных интегралов от слэтеровских функций, центрированных на разных атомах, превращается в чрезвычайно трудоемкую задачу.
Общие формулы.
Орбитали слэтеровского типа (ОСТ, англ. Slater type orbital, STO) для атомов (в сферических координатах) имеют вид:
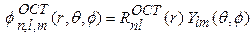 (1)
(1)
где 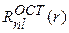 – радиальная часть:
– радиальная часть:
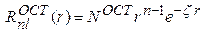 (2)
(2)
которая зависит от параметра 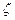 ("экспоненты"), задающего характеристики функции;
("экспоненты"), задающего характеристики функции; 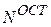 – нормировочный множитель для ОСТ (явный вид которого не приводится ввиду громоздкости формул).
– нормировочный множитель для ОСТ (явный вид которого не приводится ввиду громоздкости формул).
Орбитали гауссова типа (ОГТ, англ. Gaussian type orbital, GTO) для атомов имеет вид:
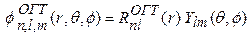 (3)
(3)
где 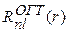 – радиальная часть:
– радиальная часть:
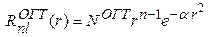 (4)
(4)
зависящая от "экспоненты" 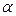 , и включающая нормировочный множитель для ОГТ
, и включающая нормировочный множитель для ОГТ 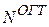 .
.
В выражениях (1) и (3) сферические гармоники 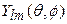 описывают угловую зависимость базисных функций. Они связанны с угловым моментом, который определяется значением l. ОСТ и ОГТ в атомах называются, как обычно, орбиталями s-, p-, d -,… типа при l = 0, 1, 2,… Вид сферических гармоник хорошо известен:
описывают угловую зависимость базисных функций. Они связанны с угловым моментом, который определяется значением l. ОСТ и ОГТ в атомах называются, как обычно, орбиталями s-, p-, d -,… типа при l = 0, 1, 2,… Вид сферических гармоник хорошо известен:
| l = 0 s | l = 1 p | l = 2 d | |
| m = -2 | 
| ||
| m = -1 | 
| 
| |
| m = 0 | 
| 
| 
|
| m = +1 | 
| 
| |
| m = +2 | 
|
При расчетах молекул применяют ОГТ частного вида, не содержащие в явном виде сферических гармоник , определяемые в декартовых координатах следующим образом:
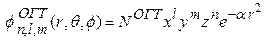 (5)
(5)
Формула (5) может быть получена при определенных дополнительных условиях из формул для ОГТ в сферических координатах (3) и (4). Система функций (5) содержит лишь функции вида 1 s, 2 p, 3 d, 4 f,…, то есть более конкретно,
s -функцию (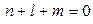 ):
): 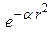 (6а)
(6а)
p -функции (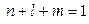 ):
): 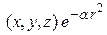 (6б)
(6б)
d -функции (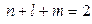 ):
): 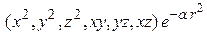 и т.д. (6в)
и т.д. (6в)
Здесь следует обратить внимание на то, что при использовании декартовых гауссовых функций в форме (5) и (6) получается шесть d -функций (6в). Лишняя составляющая (s -типа) может быть исключена посредством построения соответствующих линейных комбинаций. Аналогичная ситуация имеется также в случае функций, отвечающих более высоким значениям углового момента.
Общие выражения для разложения орбиталей по ОСТ и ОГТ имеют вид:
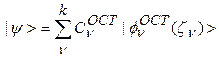 (7а)
(7а)
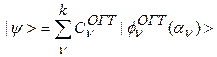 (7б)
(7б)
где индекс 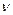 представляет собой совокупность всех индексов, задающих каждый член разложения и нумерующих члены одного типа.
представляет собой совокупность всех индексов, задающих каждый член разложения и нумерующих члены одного типа.
Сравнение ОСТ и ОГТ.
Основные различия между слэтеровскими и гауссовыми функциями могут быть продемонстрированы на примере 1 s орбитали атома водорода. Для этого следует рассмотреть 1 s орбиталь, представленную одной ОСТ и одной ОГТ (то есть, в разложении (7) содержится всего один член, k = 1):
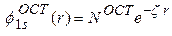 (8а)
(8а)
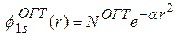 (8б)
(8б)
При 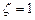 слэтеровская орбиталь (8а) является точным решением уравнения Шредингера для атома водорода, отвечающим полной энергии E = - 0.5 ат. ед. В отличие от слэтеровской функции, гауссова функция (8б) не является решением уравнения. Наилучшее значение ее экспоненты
слэтеровская орбиталь (8а) является точным решением уравнения Шредингера для атома водорода, отвечающим полной энергии E = - 0.5 ат. ед. В отличие от слэтеровской функции, гауссова функция (8б) не является решением уравнения. Наилучшее значение ее экспоненты 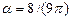 , определенное на основе вариационного принципа, дает полную энергию E = - 0.4244 ат. ед., которая заметно выше точной.
, определенное на основе вариационного принципа, дает полную энергию E = - 0.4244 ат. ед., которая заметно выше точной.
Кроме того, гауссова 1 s функция имеет неправильное поведение вблизи начала координат (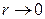 ) и на больших расстояниях (
) и на больших расстояниях (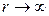 ), что видно из следующей схемы:
), что видно из следующей схемы:

Для многоэлектронных атомов слэтеровские функции перестают быть точным решением и приобретают, как и гауссовы функции, неправильное асимптотическое поведение. Кроме того следует отметить, что оба типа функций имеют неправильную безузловую структуру. Так, например, для атома лития наилучшая 2 s слэтеровская функция дает распределение электронной плотности 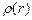 , у которого отсутствует узел в области 1 ат. ед., имеющийся в случае точного численного хатри-фоковского решения:
, у которого отсутствует узел в области 1 ат. ед., имеющийся в случае точного численного хатри-фоковского решения:
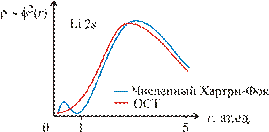
Отмеченные недостатки исправляются за счет перехода к рядам из базисных функций. Но и здесь разложения по ОСТ ведут себя лучше, чем разложения по ОГТ. При использовании ОСТ могут быть получены более точные результаты при меньшем числе членов в разложении:
Полнаяэнергия (ат.ед.) атома He (1 s 2, 1 S)
| n | ОГТ | ОСТ |
| -2.300987 | -2.847656 | |
| -2.747066 | -2.861673 | |
|
|
|
| -2.859895 | -2.861680 | |
|
|
| |
| -2.861669 | ||
| Численный Хартри-Фок (предел): -2.861680 |
(Из книги С. Фудзинага, Метод молекулярных орбиталей, М.: Мир, 1983)
В приведенной таблице содержится пример расчета полной энергии атома He, в котором варьировалось число членов в разложении 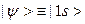 по ОСТ и ОГТ. Как видно, точный результат при использовании ОСТ воспроизводится (в пределах шести знаков после запятой) уже при пяти членах разложения. При использовании ОГТ даже десять членов разложения дают результат, который еще довольно далек от точного и сопоставим с результатом для двухчленного разложения по ОСТ. Таким образом можно лишь сожалеть, что ОСТ не подходят для молекулярных расчетов. Недостатки ОГТ компенсируют более длинными базисными разложениями.
по ОСТ и ОГТ. Как видно, точный результат при использовании ОСТ воспроизводится (в пределах шести знаков после запятой) уже при пяти членах разложения. При использовании ОГТ даже десять членов разложения дают результат, который еще довольно далек от точного и сопоставим с результатом для двухчленного разложения по ОСТ. Таким образом можно лишь сожалеть, что ОСТ не подходят для молекулярных расчетов. Недостатки ОГТ компенсируют более длинными базисными разложениями.
Подготовка (оптимизация) базисов.
В рассмотренном примере при расчете атома гелия варьировались как коэффициенты разложения { 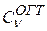 } и {
} и { 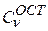 }, так и экспоненты {
}, так и экспоненты { 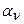 } и {
} и { 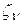 }. Такой подход однако отличается от идеологии метода Рутана, где варьируются только коэффициенты разложения.
}. Такой подход однако отличается от идеологии метода Рутана, где варьируются только коэффициенты разложения.
Вычисления с варьированием экспонент по причине трудоемкости обычно применяется только в случае атомов и только в контексте так называемой процедуры оптимизации базисов. В процессе оптимизации для каждого атома и заданной длины разложения k вариационно определяются экспоненты функций, входящих в разложения, и коэффициенты при них. После того как базис оптимизирован (подготовлен), он может использоваться в молекулярных расчетах, где в рамках метода Рутана уже варьируются только коэффициенты разложения. Оптимизация базисов – непростая задача ввиду большого числа варьируемых параметров и нелинейной зависимости АО от экспонент.
Рецепт выбора оптимальных экспонент в слэтеровских функциях был разработан Слэтером в 30-е годы прошлого века. В случае гауссовых функций наборы экспонент получали многие авторы, начиная с 50-х годов прошлого века. При этом использовалось две стратегии: (1) разложение ОСТ по ОГТ и (2) непосредственная оптимизация ОГТ.
Базисы ОГТ, полученные первым способом, как правило, являются минимальными (каждая АО описывается одной базисной функцией). В литературе они обозначаются как STO- n G (где n – число, указывающее по скольким ОГТ разложена каждая ОСТ).
Типы базисов ОГТ.
Гауссовы базисы, полученные способом (2) отличаются большим многообразием. В литературе можно найти разложения с разным количеством членов.
Для расширенных базисов (каждая АО описывается более чем одной базисной функцией) часто можно встретить следующую классификацию. Если валентные АО описываются двумя базисными функциями, то базис называется двухэкспонентным (2- 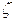 , англ. double zeta, DZ), если тремя функциями – трехэкспонентным (3- , англ. triple zeta, TZ) и т.д.
, англ. double zeta, DZ), если тремя функциями – трехэкспонентным (3- , англ. triple zeta, TZ) и т.д.
В целях сокращения числа определяемых в методе Рутана параметров члены разложения (7) иногда объединяют в группы, коэффициенты внутри которых не варьируются. При этом варьируются лишь коэффициенты при группах. Например:
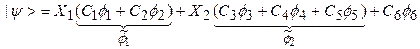 (9)
(9)
Здесь для исходного разложения из 6 членов выполнено объединение первых двух членов в новую базисную функцию 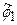 и последующих трех членов в базисную функцию
и последующих трех членов в базисную функцию 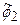 . В расчете будут варьироваться коэффициенты
. В расчете будут варьироваться коэффициенты 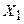 ,
, 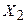 и
и  . Коэффициенты
. Коэффициенты 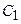 -
- 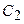 и
и 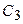 -
-  варьироваться не будут. За счет этого число базисных функций в расчете уменьшается с 6 до 3. Поскольку первые 2 функции описываются не индивидуальными ОГТ, а целыми разложениями, качество результатов при этом во многом сохраняется и оказывается лучше, чем в случае несгруппированных базисов того же размера. Неварьируемые коэффициенты - и - также как и экспоненты определяются в процессе процедуры оптимизации базисов для атомов.
варьироваться не будут. За счет этого число базисных функций в расчете уменьшается с 6 до 3. Поскольку первые 2 функции описываются не индивидуальными ОГТ, а целыми разложениями, качество результатов при этом во многом сохраняется и оказывается лучше, чем в случае несгруппированных базисов того же размера. Неварьируемые коэффициенты - и - также как и экспоненты определяются в процессе процедуры оптимизации базисов для атомов.
Базисные наборы описанного типа называются сгруппированными (контрактированными), а прием объединения членов разложения в группы – котрактацией. В настоящее время большинство базисов ГТО относятся к этому типу. Пример:
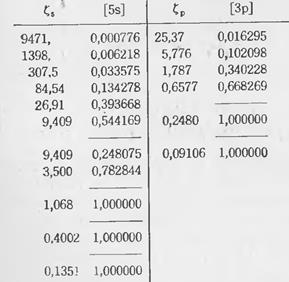
Приведенный в таблице гауссов базис (экспоненты здесь обозначены через ) для атома углерода обозначается как [5 s 3 p ], что означает, что он состоит из 5 функций s -типа и 3 функций p -типа. Квадратные скобки указывают на то, что базис контрактированный. Как видно, базис [5 s 3 p ] получен из "расконтрактированного" базиса (11 s 6 p), состоящего из 11 "примитивных" функций s -типа и 6 примитивных функций p -типа с применением схемы контрактации (группировки): (62111/411).
Если в базисный набор для некоторого атома включаются функции с более высоким значением углового момента, чем это необходимо для описания занятых орбиталей данного атома (например, d -функции в случае атома углерода), то такие функции называются поляризационными. В настоящее время установлено, что надежные результаты в молекулярных расчетах могут быть получены только при использовании поляризационных функций. Поляризационные функции улучшают описание электронного распределения в молекулах ("поляризацию атомных орбиталей" под действием молекулярного поля низкой симметрии).
В расчетах возбужденных и анионных состояний используют базисные функции, называемые диффузными. Такие функции характеризуются малыми экспонентами и улучшают описание орбиталей, являющихся вакантными в нейтральных атомах, на которые возбуждаются (или присоединяются) электроны.
В настоящее время имеется большой выбор базисных наборов. Все они опубликованы в статьях, а многие даже входят в состав квантовохимических программ. Кроме того продолжают появляться все новые базисные наборы, удовлетворяющие возрастающим потребностям расчетной квантовой химии. Современные базисные наборы обычно являются контрактированными расширенными базисами с поляризационными функциями и элементами диффузности. Их получают сразу для больших групп атомов, и они являются достаточно универсальными. Разработаны целые систематические иерархии базисных наборов, создающие возможность последовательного улучшения результатов расчетов. Примером может служить семейство корреляционно согласованных валентно поляризованных базисных наборов (англ. correlation consistent polarized valence basis sets), ставшее на сегодня своего рода стандартом современных базисов. Оно состоит из последовательности, в которую входят двухэкспонентные, трехэкспонентные, и т.д. базисы, обозначаемые как cc-pVDZ, cc-pVTZ, cc-pVQZ,... Для атомов второго периода перечисленные базисы представляют собой наборы ОГТ [3 s 2 p 1 d ], [4 s 3 p 2 d 1 f ], [5 s 4 p 3 d 2 f 1 h ],.... с общим числом функций: 14, 30, 55,... Расчеты с использованием такой базисной последовательности не только позволяют изучить сходимость результатов, но и провести их экстраполяцию к хартри-фоковскому пределу.

