 2015-04-01
2015-04-01 5343
5343Синдромы при изменении числа хромосом.
Синдром Дауна.
В качестве возможных причин синдрома Дауна рассматривались многие факторы, но в настоящие время твердо установлено, что в основе его лежит аномалия хромосом. Л
В основе синдрома Дауна лежит не расхождение одной из хромосомных пар, обозначаемой как 21-я. В результате у ребенка появляется лишняя (третья) 21-я хромосома. Это состояние называется трисомией по 21-й паре хромосом. Хотя в подавляющем большинстве случаев при синдроме Дауна обнаруживается именно эта трисомия, крайне редко встречаются и другие хромосомные аномалии.
Генетические исследования на плодовых мушках (дрозофилах) показали, что важнейшим фактором, определяющим не расхождение хромосом при созревании яйцеклетки, является возраст матери.
Однако синдром Дауна нельзя считать наследственным заболеванием, так как при нем не происходит передача дефектного гена из поколения в поколение, а расстройство возникает на уровне репродуктивного процесса.
Клиника
Симптоматика разнообразна: это и врождённые пороки развития, и нарушения постнатального развития нервной системы, и вторичный иммунодефицит и др. Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8-10% ниже средних величин). Многие симптомы синдрома Дауна заметны при рождении, в последующем они проявляются более чётко. Квалифицированный педиатр ставит правильный диагноз синдрома Дауна в родильном доме не менее чем в 90% случаев. Из черепно-лицевых дизморфий отмечаются монголоидный разрез глаз (по этой причине синдром Дауна долго называли монголоидизмом), круглое уплощённое лицо, плоская спинка носа, эпикант, крупный (обычно высунутый) язык, брахицефалия, деформированные ушные раковины.
Характерна мышечная гипотония в сочетании с разболтанностью суставов. Часто встречаются врождённый порок сердца,характерные изменения дерматоглифики (четырёхпальцевая, или «обезьянья», складка на ладони, две кожные складки вместо трех на мизинце, высокое положение трирадиуса и др.). Пороки ЖКТ наблюдаются редко. Частота какого-либо симптома в 100% случаев, кроме низкого роста, не отмечена. Большое значение для диагностики имеет динамика физического и умственного развития ребёнка. При синдроме Дауна и то и другое задерживается. Рост взрослых больных на 20 см ниже среднего. Задержка в умственном развитии достигает имбецильности, если не применяются специальные методы обучения. Дети с синдромом Дауна ласковые, внимательные, послушные, терпеливые при обучении. Коэффициент умственного развития (IQ) у разных детей широко варьирует (от 25 до 75).
Синдром Эдвардса.
 Синдром Эдвардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13.
Синдром Эдвардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13.
Этиология
Причиной заболевания является наличие дополнительной 18-й хромосомы, трех вместо двух в норме для диплоидного набора в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки —гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
Патогенез
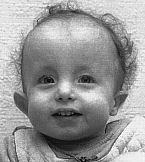
Дети с трисомией 18 рождаются с низким, в среднем 2177 г, весом. При этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Клиника
Множественные пороки развития отчетливо заметны и непосредственно обуславливают очень раннюю смертность детей с данной патологией. Их двигательная активность сильно снижена. Темпы физического и психического развития крайне низкие, и их можно проследить лишь у девочек, которые при синдроме Эдвардса способны дожить до полугодовалого возраста (очень редко – до 1 года). Мальчики же с данной патологией погибают в первые недели после рождения.
Внешние аномалии развития многочисленны и патогномоничны для синдрома Эдвардса. У ребенка подбородок скошен, челюсти недоразвиты, ушные раковины необычно низко посажены, нередко асимметричны и деформированы. Голова увеличена в продольных размерах, вместе с тем бипариетальный размер меньше нормы. Имеются множественные деформации кистей и стоп. Пальцы их обычно очень короткие (но в отдельных случаях встречаются признаки арахнодактилии). Выражены аномалии костно-мышечной системы. Мышцы с признаками атрофии, грудина и грудная клетка укорочены и деформированы. Характерно выраженное недоразвитие мышц тенара.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Синдром Патау.

Синдром Патау — это хромосомная аномалия, синдром при котором у пациента есть дополнительная 13 хромосома, в связи с нерасхождением хромосом во время мейоза (также известный как трисомия 13 и трисомия D). Дополнительная 13 хромосома нарушает нормальный ход развития ребенка, а именно возникновение дефектов сердца и почек, кроме других особенностей, характерных для синдрома Патау. Как и все болезни, которые вызваны нерасхождением (такие как синдром Дауна исиндром Эдвардса), риск возникновения этого синдрома у потомства увеличивается с возрастом матери при беременности (в среднем с 31 года). Синдром Патау поражает примерно 1 человека из 10000 новорожденных.
В большинстве случаев причиной появления синдрома Патау является трисомия 13, которая означает, что каждая клетка тела имеет три копии 13 хромосомы вместо обычных двух. Кроме того, бывают случаи (их доля очень незначительна), когда лишь некоторые клетки организма имеют дополнительную копию хромосомы в результате смешанной популяции клеток с различным числом хромосом.
В большинстве случаев синдром Патау не наследуется, а возникает как случайное событие в процессе формирования половых клеток (яйцеклеток и сперматозоидов). Ошибка в делении клеток, которая называется нерасхождением, может привести к появлению репродуктивных клеток с аномальным числом хромосом. Например, яйцеклетка или сперматозоид могут получить дополнительную копию хромосомы. Если одна из этих атипичных половых клеток будет вовлечена в генетической структуре ребенка, ребенок будет иметь дополнительную 13 хромосому в каждой из клеток организма. Мозаицизм синдрома Патау также не наследуется, а возникает как случайное нарушение при делении клеток в начале развития плода.
 Клиника
Клиника
Плод, который выживает в течение беременности и рождения, имеет следующие отклонения:
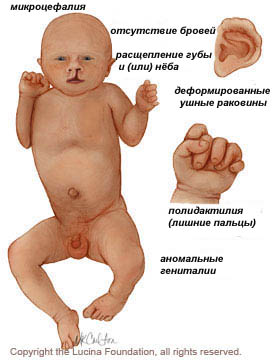
Нервная система:
- отклонения психического и моторного развития;
- микроцефалия;
- нарушение формирования полушарий мозга;
- структурные дефекты глаз, в том числе микрофтальмия, аномалия Питерса, катаракта, колоб, дисплазия или отслоение сетчатки, сенсорный нистагм, пробковая потерю зрения и гипоплазия зрительного нерва;
- спинномозговой дефект
Костно-мышечные и кожные:
- полидактилия («лишние пальцы»)
- низко посаженные и деформированные ушные раковины;
- выступающая пятка;
- деформация ноги, стопа выглядит как качеля;
- брюшной дефект, пупочная грыжа
- аномальный вид кисти;
- перекрытие пальцами большого пальца;
- врожденное отсутствие кожи (отсутствуют участки кожи / волос);
- волчья пасть, заячья губа (расщепление неба).
Урогенитальные:
- аномальные гениталии;
- дефекты почек.
Такое количество внутренних пороков быстро развивает многоорганную недостаточность. Более 90% детей погибают в течение первого года жизни. Но некоторые доживают до 5 и даже до 10 лет. Продление жизни обеспечивается оперативным устранением пороков развития, тщательным уходом, полноценным питанием. Дети с синдромом Патау всегда имеют стойкие нарушения интеллекта.
Синдром Клайнфельтера.
Синдром Клайфельтера - это заболевание, при котором особи мужского пола имеют дополнительную Х-хромосому. Это наиболее распространенное заболевание половых хромосом у мужчин и второе наиболее распространенное расстройство, вызванное наличием дополнительных хромосом. Это нарушение возникает примерно у 1 человека из 1000.
Этиология.
Появление дополнительной Х-хромосомы в кариотипе больных обусловлено нерасхождением половых хромосом у родителей или неправильным делением зиготы (оплодотворенной половой клетки). Причиной возникновения хромосомных аномалий, могут быть инфекции (особенно вирусные), немолодой возраст матери, неполноценность иммунной системы родителей и другие факторы.

